Abstract
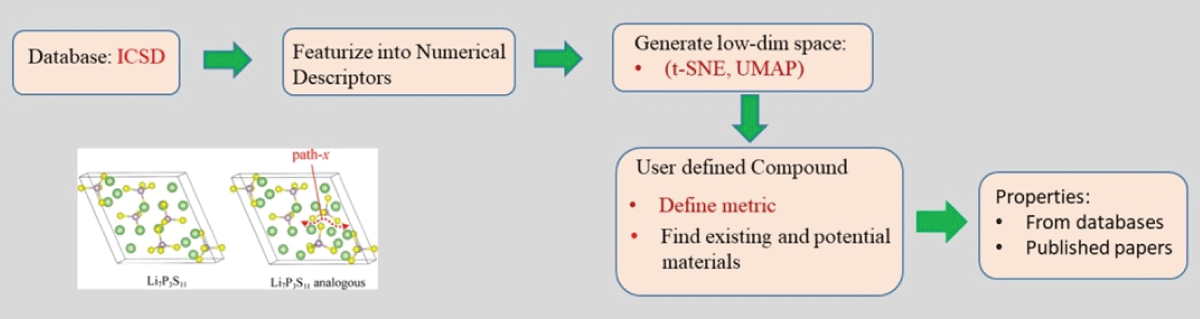
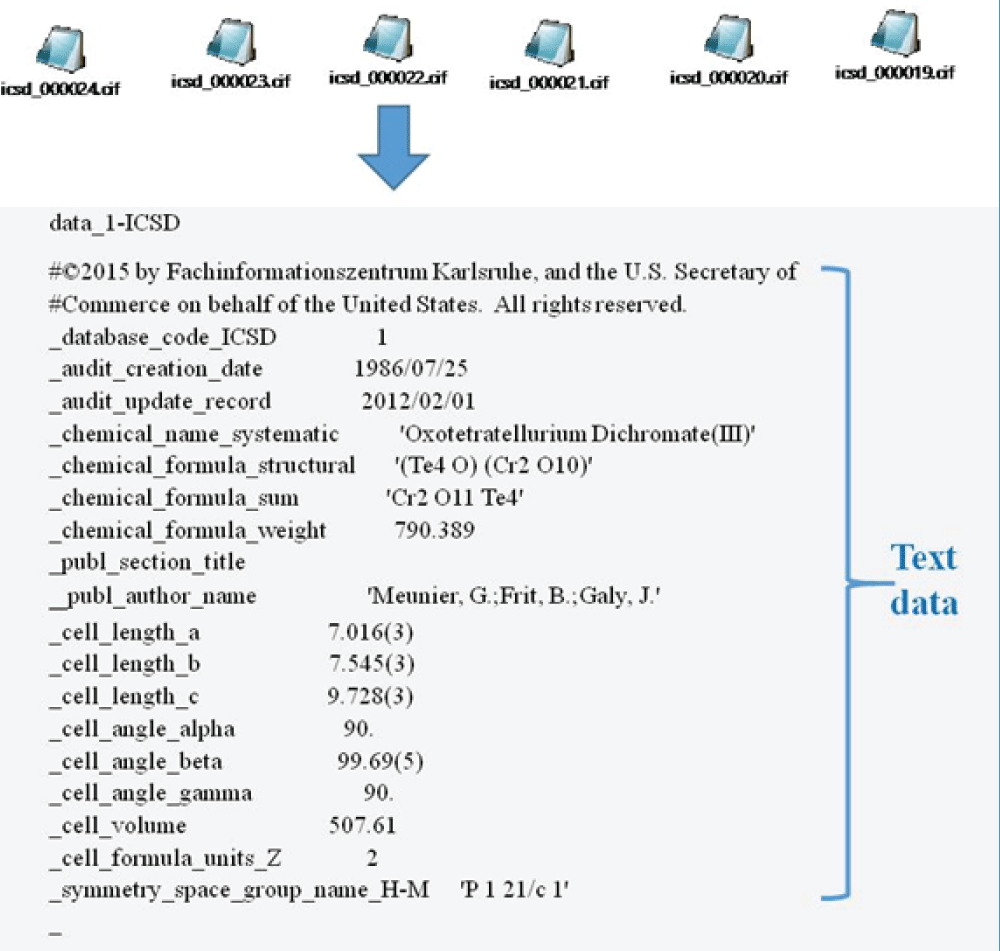
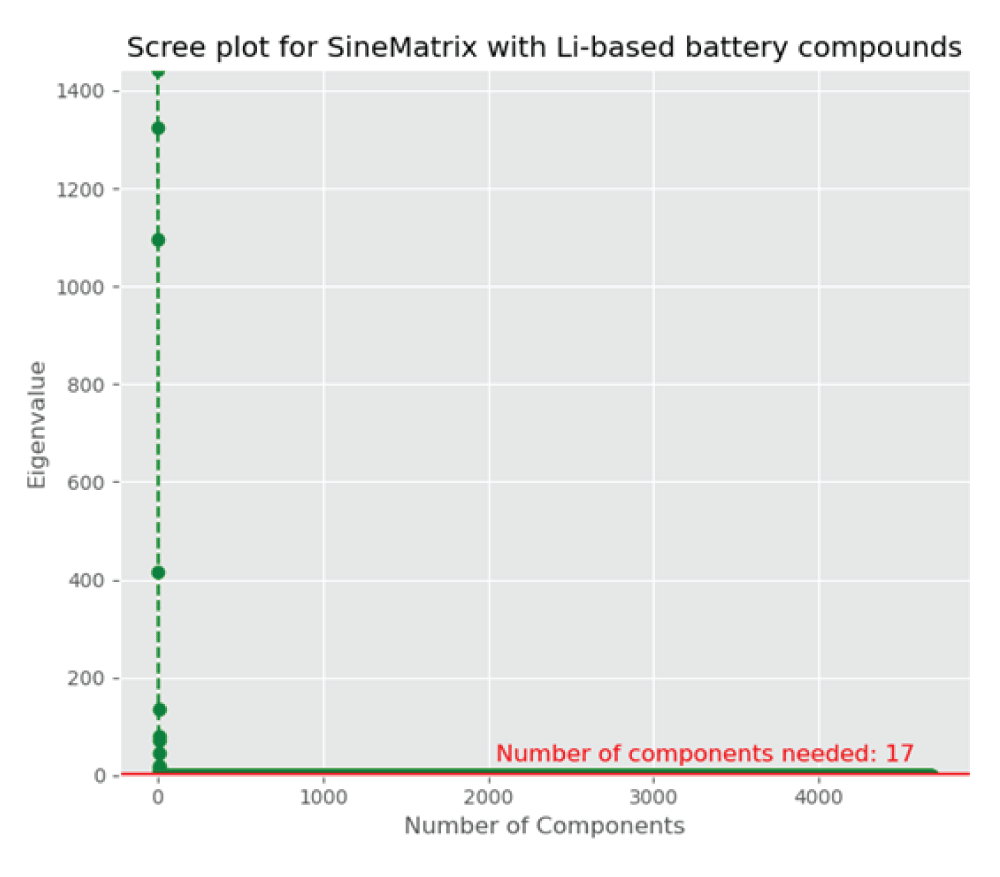
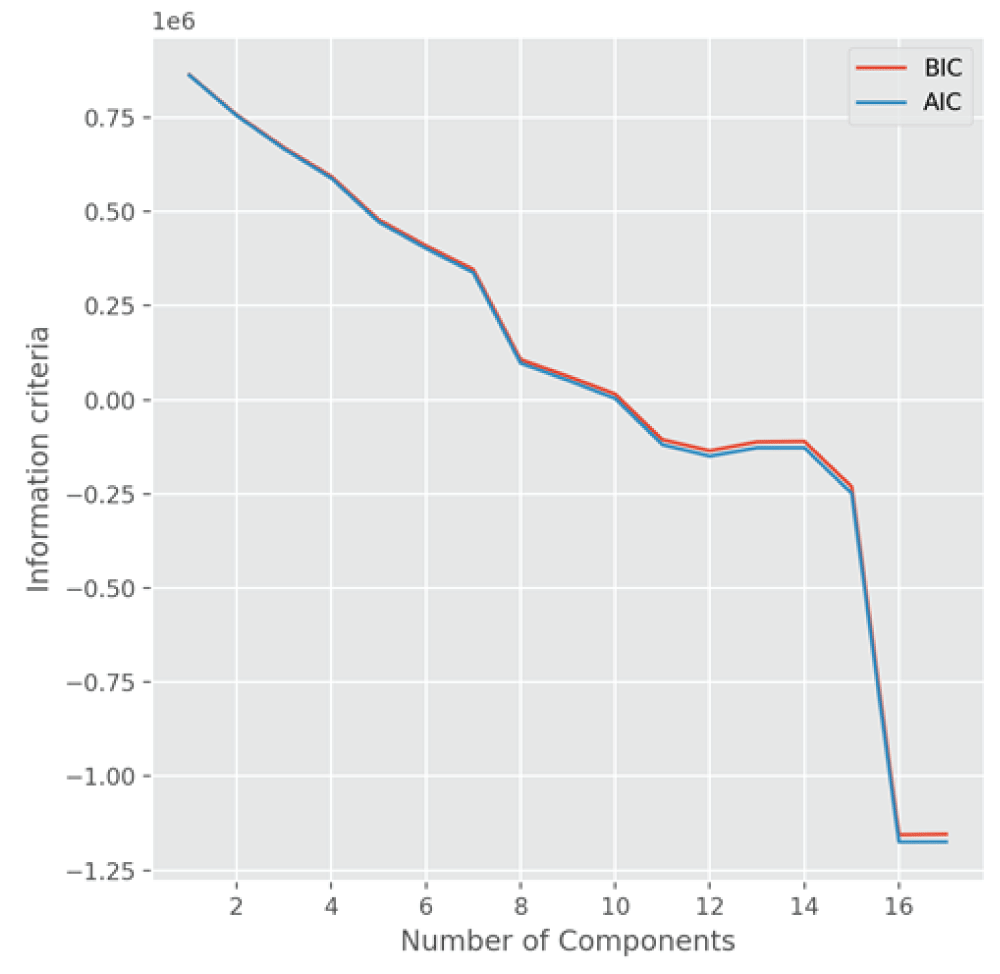
The discovery of novel functional materials is frequently accelerated by machine learning (ML) techniques that visualize vast chemical spaces in low-dimensional projections. However, projecting high-dimensional materials data into a fixed 2D space can introduce significant distortions, hindering the reliable identification of new candidates. In this work, we present a robust statistical framework for unsupervised materials discovery that mitigates these challenges. Our methodology combines Principal Component Analysis with information criteria to determine the optimal dimensionality for representing a given material's dataset, thereby minimizing information loss. We apply this approach to a library of thousands of Li-based compounds, described by their chemical and structural features. Following dimensionality reduction to the statistically optimal space, we employ a non-linear unsupervised learning algorithm to identify novel materials in proximity to a user-defined reference compound. The efficacy of our methodology is demonstrated by its ability to identify candidate materials that have been experimentally reported to exhibit properties similar to the chosen reference, validating our approach as a more reliable pipeline for accelerated materials discovery